Nature|2型糖尿病发病机制
依据2018年美国糖尿病协会(ADA)颁布的糖尿病分类和诊断标准,糖尿病可分为以下类型:
一、1型糖尿病(自身免疫性β细胞破坏,通常导致绝对的胰岛素不足,即根源少)
二、2型糖尿病(在胰岛素抵抗的条件下,β细胞分泌的胰岛素逐渐减少,即作用弱)
三、妊娠糖尿病(GDM)(在怀孕前并未明确表现为糖尿病,在妊娠中被诊断的糖尿病)
四、由于其他原因引起的特定类型的糖尿病,例如单基因糖尿病综合征(例如新生儿糖尿病),外分泌胰腺疾病(例如囊性纤维化和胰腺炎)以及药物或化学性糖尿病(例如,使用糖皮质激素,治疗HIV / AIDS或器官移植后)

不同类别的糖尿病,其病因和特征不一样。但总会涉及两个关键要素:葡糖糖和胰岛素。
胰岛素是是一重蛋白质激素,由胰脏內的胰岛β细胞分泌:
胰腺将胰岛素分泌到血液中。胰岛素循环,使糖进入细胞,进而减少血液中的糖分。随着血糖水平下降,胰腺中胰岛素的分泌也会下降。
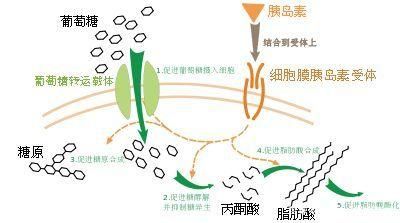
葡萄糖是一种单糖,是构成肌肉和其他组织的细胞的主要能量来源。
葡萄糖有两个主要来源:食物和肝脏。糖被吸收到血液中,并在胰岛素的帮助下进入细胞。肝脏可储存并制造葡萄糖。当葡萄糖水平低时,例如一阵子没有进食,肝脏会将储存的糖原分解为葡萄糖,以使葡萄糖水平保持在正常范围内。其中,2型糖尿病最为常见,曾经被称为成人发病型糖尿病,占糖尿病患者的9成以上。但是,目前更多的儿童被诊断出患有这种疾病,这可能是由于儿童肥胖症的增加所致。
因此,下面着重介绍2型糖尿病的发病机制。
编译 | 胡小话
2型糖尿病 (T2D) 作为当前最为常见的一种代谢疾病,在过去的50年间,患病人数持续增长,并且呈现出由西方国家向亚、非洲等西太平洋国家蔓延的趋势。另外,根据现有的模型预计,截至到2045年,全球约有将近7亿人将受这一疾病所困扰【1】。
与先天性的胰岛素分泌不足所致的1型糖尿病不同,胰岛素抵抗 (insulin resistance) 被认为是导致T2D发生发展的主要原因。虽然近些年关于T2D的研究非常之多,但具体的致病机制我们直到现在依然不是很清楚。

近日,耶鲁糖尿病研究中心联合主任Gerald I. Shulman 教授与德国糖尿病研究中心的Michael Roden教授在 Nature共同发表了题为The integrative biology of type2 diabetes的综述文章。作为研究糖尿病的顶级学者(美国科学院院士论文总被引次数超过11万 ),也是第78届班廷奖 (班廷奖作为ADA学会最高荣誉,其目的是表彰对糖尿病的认识、治疗或预防长期做出杰出贡献的医务工作者) 的获得者,Gerald I. Shulman 教授的工作对于认识和了解2型糖尿病的起源和发展非常重要。

在这篇综述文章中,Shulman教授和Roden教授通过聚焦近些年相关动物模型以及人的临床研究数据,系统总结了胰岛素抵抗和肝脏糖异生增加与肥胖以及2型糖尿病等疾病发生的内在联系。
一、肝脏脂肪之间的交流与胰岛素抵抗
在肝脏中,糖原分解 (glycogenolysis) 和糖原异生 (gluconeogenesis) 之间的稳态对于血糖的稳定至关重要。在禁食 (fasting) 过程中,胰岛素刺激的葡萄糖吸收是受损的,而血清中的支链氨基酸,游离脂肪酸含量会上升,尽管这个时候血糖浓度偏低或者趋于正常。但随着饥饿时间的延长,机体开始由葡萄糖氧化供能向脂肪酸氧化 (FAO) 供能转化,并且肝脏中糖原异生会增加。
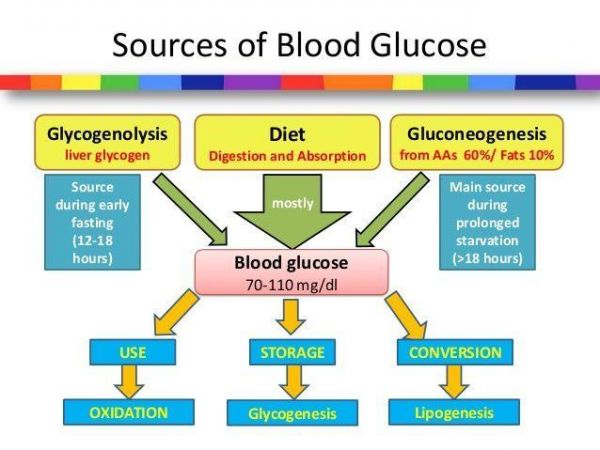
事实上,糖原异生增加在正常生理状态下应该归功于胰岛素水平的降低和糖原浓度的升高。而最近在大鼠的研究中表明:饥饿状态下,瘦素 (leptin) -下丘脑-垂体-肾上腺 (HPA) 信号轴可以通过调控白色脂肪组织 (WAT) 的脂解来介导由进食向饥饿过程的转变,这与在糖尿病人中所发现的机制是十分类似的【2】。这些研究表明,早期肝糖原分解下降是导致血清中胰岛素和血糖水平下降的主要因素,并且会使得瘦素水平下降约50%。而瘦素水平的降低则会通过HPA轴促进WAT脂解,释放游离脂肪酸和甘油,并伴随着FAO的增加。
随着更多的脂肪酸进入肝脏供给FAO,肝脏内乙酰辅酶A水平升高,从而变构激活丙酮酸羧化酶 (PC) 代谢通路,促进肝脏糖原异生和内源性葡萄糖生成 (EGP) 。此外,有研究表明,饥饿会促进肝脏甘油三脂 (TAG) 和甘油二脂 (DAG) 的累积,而这一过程会促进激酶PKCε在细胞膜上的定位,从而磷酸化胰岛素受体 (IR)Thr1160位点,抑制IR活性和胰岛素信号的传递【3】。
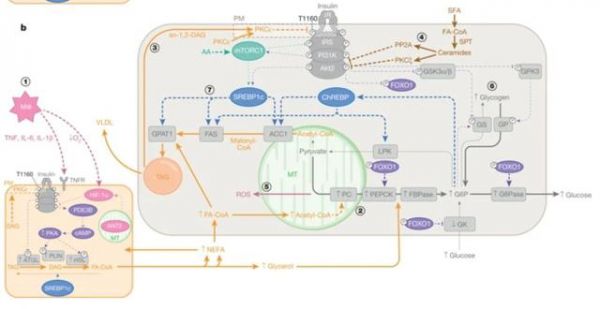
2型糖尿病患者空腹状态的高血糖,是由于肝脏胰岛素抵抗引起的脏糖原异生和EGP增加所致,而导致肝脏胰岛素抵抗的原因主要是由IR介导的肝脏胰岛素信号传递受阻的直接因素和一些其他间接因素 (包括底物可利用率、变构调节、氧化还原状态等) 所组成。而最近的研究则表明,这些间接因素可能是由于胰岛素作用于WAT所产生,并且是导致胰岛素抵抗的主要原因【4】。相比较而言,胰岛素对于肝脏直接的作用可能微乎其微,因为研究人员发现即使在啮齿类动物模型中改变肝脏中胰岛素信号的传递也并不会改变血糖的稳态。
肝脏内的胰岛素抵抗往往伴随着肝脏内TAG的累积和非酒精性脂肪肝 (NAFLD) 的发生。但是研究人员发现肝脏内的胰岛素抵抗是具有选择性的,因为只有FOXO-1介导的脂质从头合成 (DNL)受胰岛素信号调控,而SREBP1C转录调控的DNL则不受影响【5】。
这里需要注意的一点是:以上的假说必须建立在肝脏TAG来源主要是通过DNL而不是通过其他途径这一理论基础之上。而最新的一项研究显示,脂肪酸通过酯化形成TAG主要取决于游离脂肪酸转运到肝脏的效率,而与肝脏中胰岛素信号传导无关【6】。这一假说同样解释了机体胰岛素抵抗是如何导致的NAFLD的发生发展,即WAT由于胰岛素抵抗导致脂解增加并最终使得进入肝脏中的游离脂肪酸越来越多。
由此可见,肝脏与脂肪组织的相互交流与T2D患者的胰岛素抵抗密不可分。简而言之,营养过剩以及WAT功能的失调会使得WAT脂解增加,而这一过程会促进肝脏中脂质合成并导致脂肪的异位沉积以及肝糖原异生水平的上升。
二、肌肉组织中的胰岛素抵抗
肌肉组织是胰岛素的主要效应器,也是吸收葡萄糖的主力军。研究表明,对于T2D患者而言,胰岛素刺激的肌糖原合成受损是肌肉组织胰岛素抵抗的主要原因。而导致肌糖原合成下降的直接原因则是胰岛素信号通路传导受阻所致的葡萄糖转运效率的下降。T2D患者肌肉组织的胰岛素抵抗不仅会使得糖类从肌糖原合成转向肝脏,从而增加肝脏内脂质合成和甘油三脂的累积,还会使得患者体内肌肉的线粒体密度、基因表达量以及功能受损从而阻碍脂质的氧化。
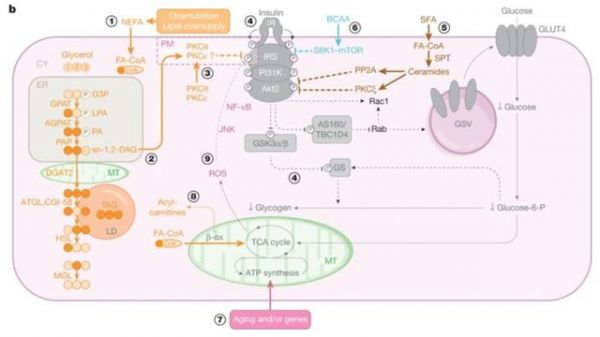
因此,这二者的叠加效应会使得肝脏进一步累积和释放TAG,并会导致肌肉内脂肪的沉积。而作者在之前已经提及:累积的TAG和TDG会通过激活PKCε抑制IR活性因此会进一步加剧肌肉组织中的胰岛素抵抗。
此外,有越来越多的证据表明,线粒体DNA (mtDNA) 以及线粒体功能相关的核DNA的多样性与T2D的胰岛素抵抗息息相关。一般来说,线粒体功能相关的核DNA的多样性往往会导致轻度的线粒体功能损坏,而mtDNA多样性则会带来更为严重的后果,如神经功能的缺陷以及胰岛β细胞的衰竭。
一项最近的欧洲GWAS分析报道,N-acetyltransferase2 (NAT2) 一个非同义突变与机体胰岛素抵抗紧密相关,并且研究人员证实在小鼠中敲低人NAT2的同源蛋白NAT1的确会导致胰岛素抵抗的表型出现,并且会导致肌肉和肝脏的脂肪异位沉积【7】。其他一些基因多样性在不改变线粒体功能的基础上同样会使得肌肉产生胰岛素抵抗,比如胰岛素信号通路中的AKT2,AS160等。
三、脂肪组织功能失调与胰岛素抵抗
与肌肉组织类似,胰岛素抵抗同样会导致脂肪组织中定位在细胞膜上IR蛋白量的减少,IR激酶活性的降低以及胰岛素刺激的葡萄糖吸收的减少。尽管WAT对于机体葡萄糖吸收的贡献不到5%,但WAT对于机体血糖的稳态却发挥着极其重要的调控作用。
原因有如下几点:
首先,WAT可以通过释放游离脂肪酸和甘油来控制肝脏糖原异生。
其次,WAT葡萄糖吸收的增加会促进ChREBP介导的脂质合成,为游离脂肪酸的酯化提供3-磷酸甘油或者产生信号分子-脂肪细胞因子(adipokines) 。
尽管脂肪重量与胰岛素抵抗存在一定的相关性,但越来越多的证据表明脂肪功能的异常更有可能是导致胰岛素抵抗的原因。有GWAS研究表明,脂肪组织的分化会影响机体的胰岛素敏感性【8】,这说明WAT储存脂肪能力的降低而不是脂肪重量的增加才是导致机体胰岛素抵抗的真正原因。
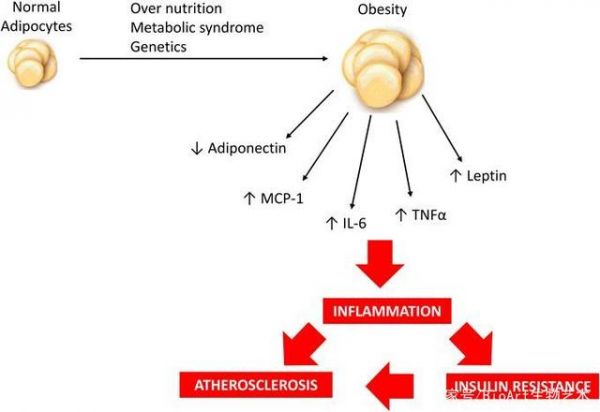
不断增加的WAT重量以及脂滴的尺寸与不充分的血管生成、低氧、纤维化以及巨噬细胞浸润导致的低烈度的炎症反应密切相关。比如说,高脂饮食和肥胖会通过激活ANT2导致脂肪细胞的局部缺氧,从而激活HIF-1α,引起脂肪组织功能失调和炎症反应【9】。此外,脂肪组织本身会释放很多细胞因子,因此,随着脂肪组织的不断增加以及伴随着各种代谢压力累积,脂肪组织释放的细胞因子则会“溢出”,从而会造成促进胰岛素敏感性细胞因子 (包括adiponectin, leptin) 和促炎细胞因子 (RBP4,resistin,IL-6以及TNF) 之间比例失衡。
但是,这里值得注意的是,胰岛素抵抗也有可能在没有炎症反应发生的情况下出现,因为研究人员分别在大鼠模型和人体内发现,高脂饮食导致的胰岛素抵抗早于炎症反应的发生之前【10,11】。
简而言之,这些发现表明代谢改变引起的脂肪异位沉积处于T2D患者胰岛素抵抗的早期,而WAT炎症反应及细胞因子的“大爆发“则会出现的更迟。最近的一些研究则表明,脂肪组织也可以通过其他途径,比如通过外泌体释放miRNAs,来调控其他组织葡萄糖耐受相关基因表达【12】。
四、大脑通过调控肝脏代谢影响机体胰岛素敏感性
事实上,除了肝脏、脂肪和肌肉这三大组织器官可以作为胰岛素主要效应器来维持机体血糖稳态,大脑对于机体的胰岛素敏感性也发挥着重要的调控作用。由于大脑具有很高的能量需求同时自身又无法存储能量,这种特殊的代谢特征使得其必须依赖葡萄糖和在饥饿状态下由肝脏产生的酮体来进行供能。
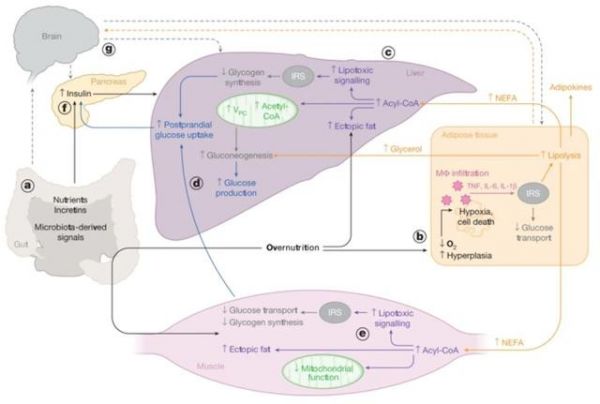
因此,大脑中的胰岛素信号或许可以通过影响食欲、情绪、认知以及外周葡萄糖代谢来发挥功能。此外,一些研究表明,大脑中的胰岛素信号会通过副交感神经促进Kupffer细胞释放IL-6来抑制肝脏的糖原异生【13】。
总的来说,大脑不仅可以通过参与不同组织器官之间的相互交流,也可以协同不同代谢物和细胞因子的相互作用,抑或是影响肠内分泌回路来发挥机体对胰岛素敏感性的调控作用。一个很好的例子就是,大脑可以在饥饿状态下通过下丘脑介导的HPA信号轴来控制WAT的脂解从而调控肝脏糖原异生和内源性葡萄糖生成【14】。
总结与展望
最后,作者对于T2D的发生发展提出了一个统一的概念。作者推测:导致空腹和餐后状态下的高血糖的根本原因是在于WAT脂解功能失调 (肝脏自身脂解异常或许也有贡献) 导致肝脏糖原异生的增加所致,而隐藏在这其中的分子机制很大可能是通过1,2-DAG-nPKC通路来的。这一概念的提出也为今后未来T2D的治疗指明了方向。
制版 | Matteo
参考文献:
1. Cho, N. H. et al. IDF Diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract.138, 271–281 (2018).
2. Perry, R. J. et al. Leptin mediates a glucose-fatty acid cycle to maintain glucose homeostasis in starvation. Cell172, 234–248 (2018).
3. Petersen, M. C. et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Invest.126, 4361–4371 (2016).
4. Perry, R. J. et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell160, 745–758 (2015).
5. Brown, M. S. & Goldstein, J. L. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab.7, 95–96 (2008).
6. Vatner, D. F. et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids.Proc. Natl Acad. Sci. USA112, 1143–1148 (2015).
7. Knowles, J. W. et al. Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J. Clin. Invest.125, 1739–1751 (2015).
8. Camporez, J. P. et al. Mechanism by which arylamine N-acetyltransferase 1 ablation causes insulin resistance in mice. Proc. Natl Acad. Sci. USA114, E11285–E11292 (2017).
9. Lee, Y. S. et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell157, 1339–1352 (2014).
10. Samuel, V. T. et al. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Invest.117, 739–745 (2007)。
11. Hernández, E. . et al. Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance. J. Clin. Invest.127, 695–708 (2017).
12. Thomou, T. et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues.Nature542, 450–455 (2017)。
13. Kimura, K. et al. Central insulin action activates Kupffer cells by suppressing hepatic vagal activation via the nicotinic alpha 7 acetylcholine receptor. Cell Rep.14, 2362–2374(2016).
14. Steinhauser, M. L. et al. The circulating metabolome of human starvation. JCI Insight3, e121434 (2018).
相关知识
2型糖尿病的运动疗法
2型糖尿病饮食建议
2型糖尿病不治能活多久
2型糖尿病的饮食治疗误区
糖尿病新药可降低心衰?2型糖尿病心衰的几率更大
2型糖尿病餐后高血糖与心血管危险
糖尿病
二型糖尿病会变瘦还是变胖
哪些糖尿病类型可治愈
全球首款一型糖尿病药物获批在即,新药可延缓糖尿病发生
网址: Nature|2型糖尿病发病机制 https://www.trfsz.com/newsview121024.html
推荐资讯




- 1男女激情后不宜做哪些事 4181
- 2从出汗看健康 出汗透露你的健 3839
- 3早上怎么喝水最健康? 3633
- 4习惯造就健康 影响健康的习惯 3283
- 5五大原因危害女性健康 如何保 3187
- 6连花清瘟、布洛芬等多款感冒药 2957
- 7男子喝水喉咙里像放了刀子一样 2455
- 810人混检核酸几天出结果?1 2225
- 9第二轮新冠疫情要来了?疾控中 2219
- 10转阴多久没有传染性?满足四个 2163

