下一代循证医学

最近,可穿戴技术、数据科学和机器学习的进步已经开始改变循证医学,为下一代“深度”医学的未来提供了诱人的一瞥。尽管基础科学和技术取得了惊人的进步,但主要医学领域的临床转化仍然滞后。药物开发与临床试验的成本和失败率居高不下,超过三分之二的化合物在实验室到临床治疗这条“死亡谷”中死去,将一种药物成功投入临床的花费超过15-25亿美元,再加上医疗系统固有的低效和缺陷,导致临床研究出现危机。
虽然COVID-19大流行暴露了临床试验领域固有的系统局限性,但它也推动了一些积极变化,包括新的试验设计,以及向更以患者为中心和更直观的证据生成系统的转变。在不久未来,机器学习、深度神经网络和多模态生物医学人工智能(AI)的应用将从各个角度重振临床研究。近期,美国MD安德森癌症中心的Vivek Subbiah教授Nature Medicine上发表了一篇文章,分享他对未来临床试验和循证医学的看法,并就临床试验的设计、实施和证据生成方面需要改进的地方进行了深度思考。魔方在此整理以供参考。

临床试验设计
随机对照试验(RCT)的挑战
RCT一直是所有医学领域产生证据的金标准,因为它们允许在没有混杂因素的情况下对疗效进行无偏倚估计。理想情况下,每种药物或干预都应通过RCT进行测试,但是,很多情况下开展RCT并不可行,原因包括及时生成证据的挑战、费用、针对较窄人群的设计限制了普适性、伦理问题以及开展试验所需的时间。当RCT完成和发表时,它们可能很快就过时了或不能适用于当下背景。仅在心脏病学领域,由于招募方面的挑战,就有30000项RCT尚未完成。此外,目前试验的设计多是孤立的,许多临床问题仍未得到解答。因此,传统的试验设计范式必须适应当代基因组学、免疫学和精准医学的快速进展。
临床试验设计进展
在过去十年中,主方案(master protocols,适用于几个子研究的总体研究方案)的设计与实施取得了实质性进展,大大改善了RCT的停滞状态。此外,主方案可能涉及对单一疾病或由生物标志物或疾病实体定义的多种疾病开展平行干预性研究。主方案包括四类不同的研究——伞形试验(umbrella study)、篮子试验(basket study)、平台试验(platform study)和主观察性试验(master observational tria,MOT)。这些研究设计都非常独特且具有灵活性。由于基因组学、新疗法和临床转化的快速发展,肿瘤学领域比其他医学领域更为领先,从而开创了精准肿瘤学时代。
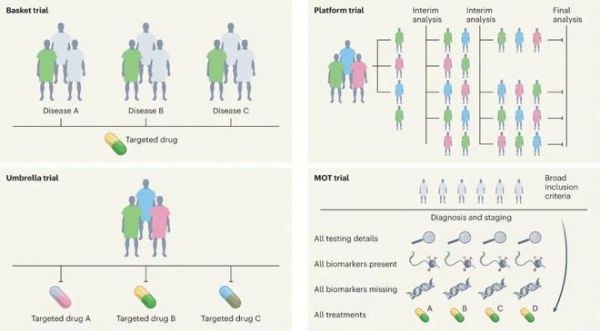
图:主方案的分类
伞形试验,是根据分子改变分层,评估针对同一疾病实体的多种靶向治疗的研究设计。如I-SPY(Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging And Molecular Analysis)乳腺癌试验和Lung- MAP(Lung Cancer Master Protocol)试验
篮子试验,是一项组织未知或与组织学无关,针对具有共同分子改变的多种疾病类型评估相应靶向治疗的研究设计。如VE-BASKET试验、ROAR(Rare Oncology Agnostic Research)试验、ARROW试验和LIBRETTO-001试验。
平台试验,是一种多组、多阶段的研究设计,在相同的主方案背景下,将几个干预组与一个共同的对照组进行比较。此外,因为没有固定的结束日期,平台试验可以一直随着时间的推移而进行,并且由于采用了共同的对照组,从而确保纳入干预/实验组的患者比例高于对照组,因此比传统试验更有效。如COVID-19治疗随机评估(RECOVERY)平台试验和英国晚期乳腺癌血浆分子谱分析指导治疗选择(plasmaMATCH)平台试验。
主观察性试验,是一项前瞻性、观察性研究设计,广泛接受不依赖于生物标志物标签的患者,并收集每名参与者的全面数据。MOT结合了主干预试验和前瞻性观察性试验设计,并试图将基于生物标志物的主干预方案的功效与真实世界数据(RWD)的广度相结合,非常适合于收集许多专业的前瞻性RWD。如肿瘤检测和治疗结果注册(ROOT)MOT。
生物标志物的开发和试验终点的定义
为了加快药物开发和临床试验,我们需要定义生物标志物(临床、病理或生理)及其在每种疾病过程中的使用背景,并为研究选择明确的终点。生物标志物可用于诊断、预后或预测,并可为早期药物开发、剂量选择和试验设计提供信息。此外,生物标志物有助于加速基础科学和药物发现,所有这些最终目标都是改善患者健康。然而,生物标志物的证据水平在很大程度上取决于使用的背景。
除了生物标志物之外,每个领域都需要明确最优先研究的问题,并确定最相关的终点,以回答所定的假设。终点是健康和/或疾病的衡量指标,根据试验阶段的不同,其作用有不同的目的。除了临床和监管终点之外,患者报告结局和数字终点(在患者日常生活的背景下,在临床环境之外收集的传感器生成的数据)也在迅速出现。
临床状况的数字化和评估需要统一和标准化,需要跨学科合作和监管投入,对于慢性疾病的中间终点和替代终点也需要达成共识。这要求除功能这一层面的数据之外,还根据专业具体情况纳入多个层面的数据,如基于基因组、蛋白质组学和基因型-表型的临床数据以及疾病特异性检测指标。美国国立卫生研究院(NIH)和美国食品药品监督管理局(FDA)开发了BEST (Biomarkers, Endpoints and other Tools)资源,以理清生物标志物和终点的不确定性,这是一个“living document”,随着标准和证据的变化而不断更新,它澄清了重要的定义,并描述了术语之间的一些层次、联系和依赖关系。
临床试验的实施
临床试验实施的组成部分包括方案的实施;患者筛选、招募、监测和保留;确保遵守安全报告;持续审查和数据分析。制药业和卫生保健部门为临床试验投入了大量资源,但迫切需要做出改变,使这一过程更加无缝。此外,临床试验的进行速度太慢了,无法与各个领域的研究进展相匹配,因此,需要对每个流程基于科技逐步进行改造。此外,高影响力、高效的研究中心只是少数,大多数临床试验中心都面临人员配备限制和其他障碍的挑战,这也是临床试验实施面临的一大现实问题。
这次COVID-19大流行的一个积极方面是,它迫使系统比以前更以患者为中心,从而更加重视临床研究的主要主题—患者。这催生了去中心化试验;数字、远程和“虚拟”试验;以及“家庭医院”和在家监测这些概念。采用基于AI的方法来增强患者体验可以进一步改善患者的实际体验评估,并确保对方案的依从性。虽然数字化、虚拟化和去中心化并不能彻底解决临床研究危机,但它们可以提高效率,对下游可能产生巨大且长期的影响。
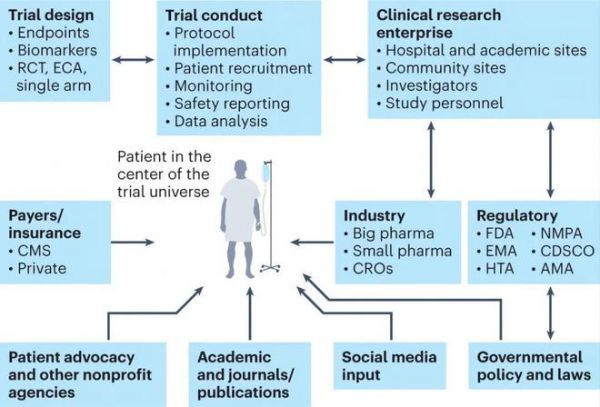
图:在临床研究产业中,患者是临床试验的中心
临床试验研究产业
临床试验研究产业的效率和协作是临床试验成功的主要因素。产业的主要组成是患者、学术中心、申办方(大小制药公司)、政府/合作团体申办方、监管机构、患者倡导组织和合同研究组织(CROs),所有这些都需要以患者为中心进行临床试验。此外,整个系统需要进行数字化改革,注册临床试验需要通过严格的电子数据采集和监测进行日常管理。可以想象,将区块链技术集成到临床试验管理系统可增加人们对临床试验过程的信任,并起到促进监管的作用。
其次,临床试验组织者还应使患者更容易参与试验,重大疾病的医患治疗决策应将临床试验方案也作为一种标准治疗。这些临床试验应易于获得,并应确保没有患者被不必要地排除在外,这一点可以通过与研究中心无关的临床试验匹配和导航服务来实现。最后,临床试验培训应成为医学教育的一部分,以便为临床研究提供经过培训的不同研究人员和工作人员。
药物开发时间表
从申请专利到FDA最终批准,候选药物的临床开发时间表是一场与时间的赛跑,药物开发时间平均约为10年。虽然FDA设立了多个项目来缩短上市前流程的时间,包括加速批准、优先审评认定、快速通道认定、突破性治疗认定和孤儿药认定,但是时间依然还是太长了,我们迫切需要解决所有疾病的这一问题,因为药物开发速度对患者、医生和药物开发利益攸关方都至关重要。
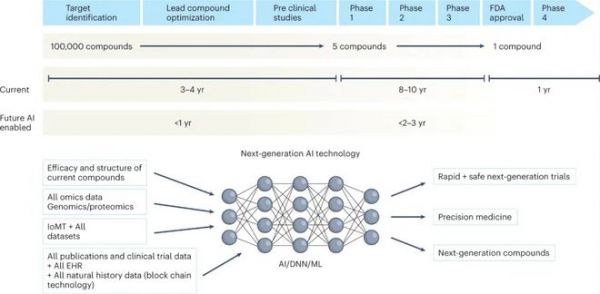
图:从现在到未来的药物开发时间表
药物开发、监管和可迁移性分析全球化
迫切需要全球各监管机构协调一致,以解决在获得药物方面的严重不平等问题。理想情况下,新疗法的临床试验应在全球范围内进行,以便患者可及和企业推广。然而,现实情况是,不可能在每个国家进行包括RCT在内的临床试验,以产生针对该国人群的特定证据。利用可迁移性分析(transportability analysis)产生的证据正在获得关注,指的是从一个国家的研究样本得出推论可以推广到另一个未开展研究国家的目标人群。可迁移性分析可能提供一些外部有效性的证据,并对当地监管和卫生技术评估产生影响。
临床试验的证据生成
罕见病的临床研究
由于疾病的罕见性和不完整的自然史数据使得罕见病的临床试验颇具挑战。然而,分子生物学的快速进展以及孤儿药法案的颁布推动了这一领域的进展。使用加速审批程序等项目的监管灵活性越来越大,在某些情况下,试验允许使用基于真实世界数据(RWD)的外部对照组(external control arms)。例如,FDA加速批准alpelisib用于2岁及以上患有PIK3CA相关过度生长谱系且需要全身治疗的成人和儿童患者,就是基于单臂EPIK-P1研究的RWD评估疗效。
此外。在个体化基因组时代,单病例随机对照试验(N-of-1 trial)正成为研究潜在致命罕见疾病的工具。还有,特定患者的医学模拟“digital twins”概念也是一个新兴的研究领域,它有可能结合多项数据如机制数据、病史,以及AI的效能,并且可能在未来强化N-of-1 trial,从而进一步实现医疗个性化
RWD和真实世界证据(RWE)
RWD是指从患者的常规标准治疗中产生的数据,而RWE是指从RWD中产生的关于产品潜在用途的证据。RWE由试验设计或分析产生,不限于随机试验,而是来自实效性试验以及前瞻性和/或回顾性观察性研究。
对所有临床试验研究的主要批评之一是,临床试验不能代表“真实世界”的人群。临床试验的限制性标准和为回答特定问题而进行的有限分析可能不适用于真实世界的患者。传统试验的设计是基于一种误解,即监管机构可能无法接受来自RWD的更现代、更多样化的证据,但现在情况已不再是这样。监管机构通过《21世纪治愈法案》为所有利益相关方提供了RWD和RWE相关的指导和全面的框架。此外,FDA使用RWD和RWE进行上市后安全性监测,保险机构也已经开始使用这些数据进行保险覆盖范围的决策。
来自合成对照组(synthetic control arm)或外部对照组的证据
罕见疾病基因组学的进步和罕见癌基因驱动的癌症的发现使得产生了特定的靶向疗法,而在RCT中对这些疗法进行评估可能不可行,也不符合伦理,并且可能推迟患者获得有希望或挽救生命的疗法。在这种情况下,合成对照组可以作为“模拟”RCT对照组的选择。合成对照组与研究无关,大多来自RWD,也可由既往临床试验(单一或合并试验)数据生成。这是一个为创新做好准备的新兴领域,因为我们可以从多个来源获得大量的数据。
使用合成对照组可以加速药物开发,最初对它们的怀疑主要源于缺乏监管机构的优先考虑和指导。随着合成对照组逐渐被用于超罕见疾病的药物审批,这些担忧正在消除。尽管取得了些进展,外部对照组仍然是一个新概念,它们主要用于调查疾病的自然史,通常未被作为主要证据或列入产品标签。然而,在未来,我们可以设想这样的疗效比较分析和对照组作为支持药物批准的主要证据。可能的挑战主要来自数据质量和数据缺失,以及不确定外部对照数据是否适用。然而,其中一些问题可以通过定量偏倚分析和其他方法来缓解。
儿科临床试验
儿科治疗新药开发方面的创新常常是滞后的,许多罕见病主要发生于儿科人群,针对这一人群的药物开发在实施、伦理、统计学和方法学方面一直具有挑战性。对基础生物学、疾病本身以及药物的急性和长期安全性的理解有限加剧了这一问题。此外,在临床试验不可行的情况下,药物在极年幼儿童、婴儿和新生儿中有相当多的超说明书使用,因此,迫切需要通过创新的方法产生高级别证据。2002年《儿童最佳药物法案》和2012年根据《FDA安全与创新法案》永久生效的《儿科研究公平法案》等项目激励儿科治疗的发展。创新的试验设计、RWD和利用来自其他资源的数据可能有助于儿科治疗的风险-获益评估和药物批准。
重新设想临床试验的未来
AI
AI有潜力增强从药物设计到整个药物开发周期的所有阶段。但目前,大多数AI研究关注的是“临床医疗服务”应用,而不是“临床试验研究”。AI与临床试验研究的整合比预期的要慢,主要是由于人工智能与人类智能之间感知的差距。使用AI的下一代临床试验应考虑AI+人类,而不是AI vs人类的场景。
目前的循证医学金字塔只是冰山一角,仅能提供治疗普通患者的浅层证据。因此,需要对所有可用数据进行深度综合和融合,才能实现下一代“深度”循证医学。未来20年的主要挑战将是通过提取、整理和挖掘大型自然史数据集、基因组学和所有其他组学分析、所有已发表的临床研究、RWD、无处不在的智能设备数据以及医疗物联网(IoMT)收集的数据来挖掘多维证据生成的潜力,为深度医学提供下一代证据。
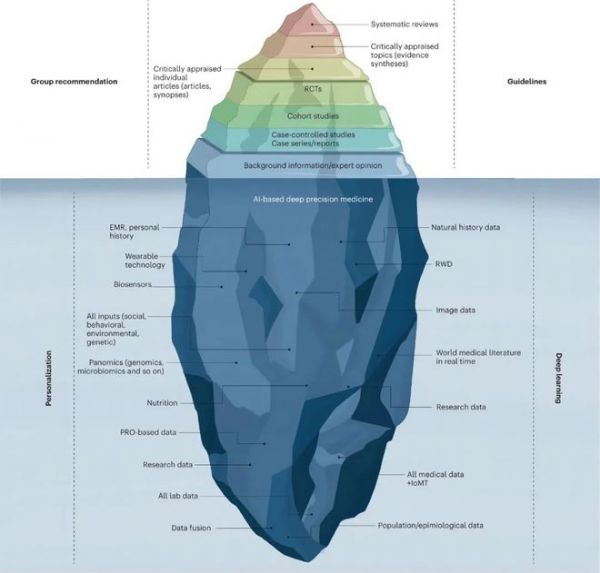
图:循证深层医学冰山
药物开发伙伴关系
目前,制药行业是药物开发的主要驱动力,他们的支出远远超过了任何国家机构的投资。在这个国家机构资金减少的时代,药物开发需要更多的伙伴关系。许多学术机构正在与制药行业建立风险分担战略联盟,在临床前和临床开发阶段进行合作。这种成功的创新伙伴关系模式在囊性纤维化、多发性骨髓瘤、1型糖尿病和其他罕见疾病中开创了先例。这些合作有效地促进了药物开发各个阶段的创新,并为维持和促进更多此类项目提供了令人信服的理由。
社交媒体和在线社区研究
社交媒体(Twitter、Facebook等)可以影响临床试验中的患者数量。它们可以极大地影响和解决以往的临床试验挑战,包括患者和医生对现有试验缺乏认识,以及缺乏社区参与。患有绝症的人经常自己尝试药物,而在线患者社区可以提供共享和监测药物使用的环境。这使得我们可以围绕基于互联网的定量结局数据规划观察性研究。增加患者和患者倡导团体的参与,有助于对患者进行教育和推广,促进与患者合作的研究,并允许在临床研究设计中纳入患者的观点,最终产生的研究是由所研究疾病的真实患者的需求驱动的。此外,社交媒体打破了研究者和临床医师之间的隔阂,创造了影响医学所有领域的巨大潜力。
总结
未来临床试验的成功需要从根本上转变试验的设计、实施、监测、调整、报告和监管方式,以产生最佳证据。临床研究当下的模式是不可持续的,我们需要预防性、个性化、务实和有患者参与的医疗,并且需要通过可持续增长来实现范式转变。方法学的进步和未来基于人工智能的所有数据分析将为实现个性化医疗的目标提供深入的证据,也就是要在正确的时间为正确的患者提供正确的治疗。
参考资料:
[1]Subbiah V. The next generation of evidence-based medicine. Nat Med. 2023;29(1):49-58. doi:10.1038/s41591-022-02160-z
Copyright © 2023 PHARMCUBE. All Rights Reserved.
欢迎转发分享及合理引用,引用时请在显要位置标明文章来源;如需转载,请给微信公众号后台留言或发送消息,并注明公众号名称及ID。
免责申明:本微信文章中的信息仅供一般参考之用,不可直接作为决策内容,医药魔方不对任何主体因使用本文内容而导致的任何损失承担责任。
相关知识
《中国全科医学》医学循证|急性痛风性关节炎治疗:针灸OR西药?
散寒化湿颗粒与新冠神药Paxlovid头对头研究引关注,中医药取得全球关注的循证医学证据
坚持科学循证,打造高质量益生菌产品
第一版循证的儿童成长里程碑发布,推荐每位家长收藏
《中国儿童食物过敏循证指南》正式发布
中西医结合、补充和替代医学概述
中医药治疗新冠病毒感染高级别循证研究结果发布
中国新生儿疼痛管理循证指南(2023年)
中医药治疗新冠最高级别循证研究成果公布!散寒化湿颗粒临床优势获力证
中医育儿的重要性:培养健康与智慧并重的下一代
网址: 下一代循证医学 https://www.trfsz.com/newsview315751.html
推荐资讯



- 1从出汗看健康 出汗透露你的健 3963
- 2早上怎么喝水最健康? 3743
- 3习惯造就健康 影响健康的习惯 3379
- 4五大原因危害女性健康 如何保 3282
- 5连花清瘟、布洛芬等多款感冒药 3006
- 6补肾吃什么 补肾最佳食物推荐 2517
- 7男子喝水喉咙里像放了刀子一样 2510
- 810人混检核酸几天出结果?1 2267
- 9第二轮新冠疫情要来了?疾控中 2260
- 10转阴多久没有传染性?满足四个 2207

