近年来,由于对神经系统疾病遗传病因的研究和基因测序技术的普及,基因检测在神经学临床实践中的应用迅速增加。此类检测现已广泛适用于成年人,尤其是患有各种常见神经系统疾病的患者。基因诊断的潜在临床影响不断扩大,包括基因特异性治疗和临床试验,对预后、监测、家庭规划和诊断等方面产生重要影响。
近期《Journal of Neurology》发表了一篇针对成人神经系统疾病的实用性临床指南。该文章着重回答了临床神经科医生的三个问题:哪些患者需要进行基因检测?应该进行哪些检测?基因检测如何帮助患者?

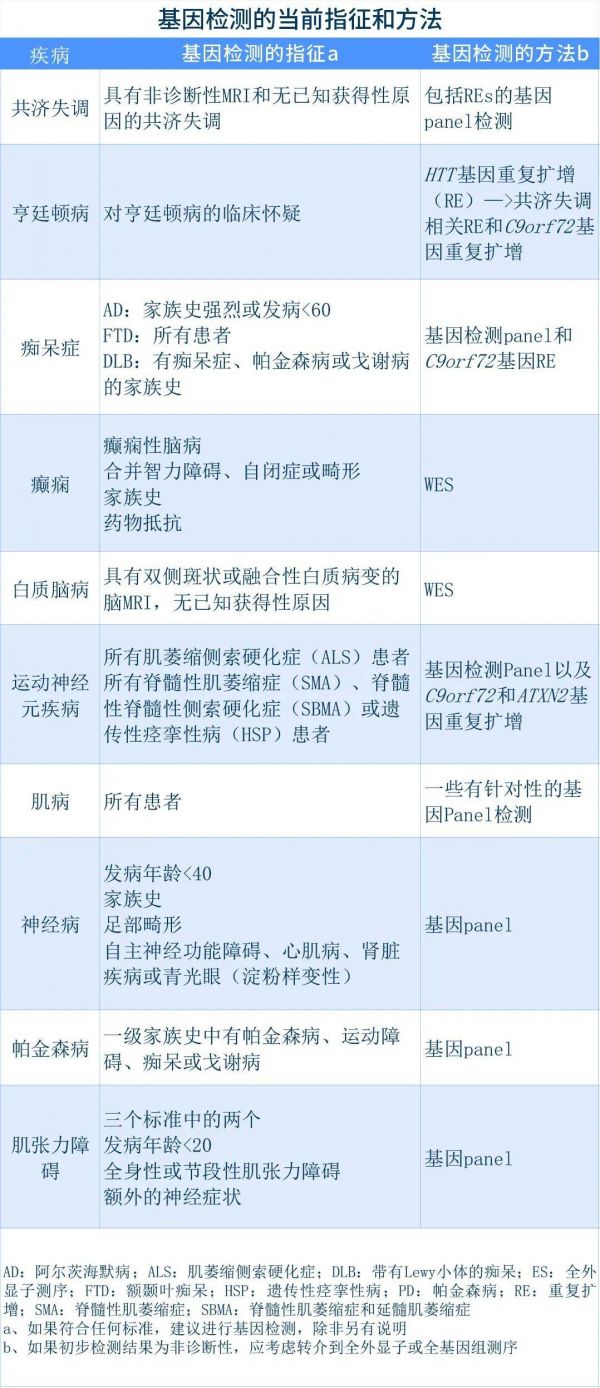
小编总结了文中介绍的神经科临床中常见的神经系统疾病的上述三个问题,并汇总成以下两个表格,此外根据文献内容展开提供更详细的介绍,以期为临床医生提供相关帮助。

以下根据文献内容针对部分疾病进行展开介绍,包括:
一、共济失调(Ataxia)
二、亨廷顿舞蹈病(Huntington’s Disease)
三、痴呆(Dementias)
四、癫痫(Epilepsies)
五、脑白质营养不良(Leukodystrophies)
六、运动神经元病(Motor Neuron Disease)
七、肌病(Myopathies)
八、周围神经病(Neuropathies)
九、帕金森病(Parkinson's Disease)
十、肌张力障碍(Dystonias)
一、共济失调(Ataxia)
基因检测的指征和阳性率:
对于患有慢性或进行性共济失调、脑部成像未提供诊断性信息或排除可逆性共济失调原因的患者,建议进行基因检测,无论其家族史如何。发病年龄较早和家族史阳性会增加基因检测的阳性率,但缺乏共济失调家族史并不排除基因原因。特定的临床特征也可能增加阳性率,阳性率范围从20%-50%不等。
检测方法:
许多成年发病的共济失调由三核苷酸重复扩增(REs)引起。建议首选有针对共济失调RE障碍的靶向panel检测,尤其是存在家族史的情况下。如果RE检测未能显示结果,考虑进行全外显子测序(WES)或全基因组测序(WGS),包括线粒体基因组。
临床进展:
对于Friedreich共济失调,Nrf2激活剂Skyclarys(omaveloxolone)已获批准,并在临床试验中显示改善神经功能。其他遗传性共济失调目前缺乏改变疾病进程的治疗方法,但核苷酸为基础的治疗方法仍在积极研究中。部分患者需要监测额外的神经症状,例如心肌病和癌症筛查。
二、亨廷顿舞蹈病(Huntington’s Disease)
基因检测的指征和阳性率:
对于亨廷顿病(HD)的基因检测,成年人中出现缓慢进行性的运动功能障碍、认知衰退和精神症状时应考虑,不论有无家族史。家族史在确定基因诊断时尤为重要,在没有已知家族史的情况下,亨廷顿病的基因检测有助于排除其他遗传性神经退行性疾病。有症状的家族成员可通过基因检测明确诊断。当临床根据表征怀疑HD时,其分子诊断率通常超过80%。
检测方法:
包括HTT基因中的CAG重复检测,亨廷顿病通常CAG重复数≥40次。如果检测为阴性但怀疑存在基因原因,可以考虑进行全外显子测序(WES)/基因组测序(WGS)检测。
临床进展:
目前FDA批准用于治疗亨廷顿病的唯二药物是丁苯那嗪和氘丁苯那嗪。
三、痴呆(Dementias)
基因检测的指征和阳性率:
针对阿尔茨海默病(AD),建议对具有强烈家族史或年轻发病(<60岁)患者进行基因检测。早发AD患者的遗传检出率为5-13%。对无家族史的晚发AD患者(>65岁)通常不建议检测。额颞叶痴呆(FTD)患者建议进行基因检测,尤其是有深部神经肌肉症状或强烈家族史的情况。当有患者有痴呆、PD或戈谢病的家族史时,应考虑进行基因检测,因为这些疾病之间存在相似的遗传风险。
检测方法:
建议首先进行C9orf72 RE检测和其它与痴呆相关的基因。初步检测结果为阴性时,可转向WES/WGS检测。怀疑存在朊病的情况可能需要进行额外检测(凝胶电泳、等位基因特异性Sanger测序或长读取测序),因为八肽重复插入通常无法通过二代测序检测到。
临床进展:
尽管目前没有获批用于基因治疗的药物,但基因检测可以提供早期和更准确的诊断。因为临床诊断并不总是与病理学一致,且目前缺乏临床可用的疾病特异性生物标志物,因此基因检测是患者明确分子诊断结果的唯一手段。
四、癫痫(Epilepsies)
基因检测的指征和阳性率:
2022年国际抗癫痫联盟基因学委员会建议对癫痫患者进行基因检测,当患者具有以下情况之一时:(1)阳性家族史;(2)智力障碍、自闭症或畸形;(3)药物耐受。如Lennox–Gastaut综合症、家族性癫痫等癫痫性脑病遗传检出率最高(>20%)。特定癫痫表型,如结节性硬化症、Dravet综合症等,由明确的遗传基因引起。
检测方法:
癫痫的遗传异质性要求采用广泛的检测策略,WES是推荐的首选检测。家系WES检测有助于解释和发现新发变异。
临床进展:
癫痫中的基因诊断可导致12-80%的病例管理变化,包括药物选择、老药新用、参与临床试验、筛查非神经系统并发症和提供预后信息。精准治疗的发展将进一步影响癫痫患者的临床管理和治疗决策。
五、脑白质营养不良(Leukodystrophies)
基因检测的指征和阳性率:
成年发病的遗传性白质脑病很难与类似多发性硬化等获得性白质病变区分开来,通常从家族史、发病年龄较小(<50岁)和多系统病变的角度判断遗传性病因。基因检测在成年患者中的阳性率为25-38%。
检测方法:
考虑到遗传性脑白质病的表型、基因的巨大异质性,倾向于广泛采用WES或WGS,并结合血清、血浆和尿液代谢检测。
临床进展:
遗传性白质脑病的基因诊断对疾病修饰治疗、监测具有重要临床影响,例如2022年FDA批准的肾上腺脑白质营养不良男孩的基因治疗(Skysona)。
六、运动神经元病(Motor neuron disease)
基因检测的指征和阳性率:
肌萎缩性侧索硬化症(ALS)的遗传检测逐渐成为新诊断患者的标准。C9orf72重复是欧洲人群最常见的遗传病因。目前已知有40多个与ALS发病机制有关的基因,总体上解释了70%的家族性ALS。对于其他运动神经元疾病,如遗传性痉挛性截瘫、脊髓性肌肉萎缩症和脊髓球部肌肉萎缩症,也应进行遗传检测。
检测方法:
2023年的共识指南建议为ALS患者提供C9orf72 RE分析和基因panel检测,ATXN2 RE检测同样重要,如果初步检测结果不明确,可进行WES/WGS检测。
临床进展:
目前,FDA已批准了针对SOD1基因的ASO药物tofersen,其他治疗方法正在研发中。对于脊髓性肌肉萎缩症患者,FDA和EMA批准的治疗方法使疾病自然史发生了有意义的变化。当前存在基因介导治疗的临床前研究,但针对SBMA的批准治疗尚未面世。
七、肌病(Myopathies)
基因检测的指征和阳性率:
遗传性肌病已确定200多个致病基因,无论家族史如何,建议所有原因不明的肌病患者进行基因检测。对于无免疫调节疗法反应的获得性肌病患者,基因检测同等适用,检出率在30-50%之间。
检测方法:
首选基因panel,不明确时进行WES/WGS。一些肌病如杜氏肌营养不良症等由复杂遗传变化引起,推荐有针对性检测。怀疑遗传性肌病可在肌肉活检前进行基因检测,减少侵入性。
然而,一些常见肌疾的遗传变化较为复杂,如肌强直性肌营养不良、Duchenne型和Becker型肌营养不良、面肩肱型肌营养不良和眼咽型肌营养不良。这些疾病可能涉及WES无法检测到的缺失或重复扩增(REs)。由于肌病的临床表现高度相似,因此在选择检测策略时,建议优先采用WES/WGS,若未能发现异常,再考虑进行REs的有针对性检测。
临床进展:
在遗传性肌肉疾病领域,最近获批的Eteplirsen和Viltolarsen已用于治疗Duchenne型肌营养不良(DMD)。庞贝病方面,早在2010年就已开始使用酶替代疗法。此外,还有多种类型的遗传性肌病正在进行靶向治疗的临床试验。
八、周围神经病(Neuropathies)
基因检测的指征和阳性率:
Charcot–Marie–Tooth病(CMT)是涉及运动和感觉神经的遗传性疾病,表现为多样化的症状。针对典型CMT表型,即远端运动无力、轻微感觉症状和足部畸形,强烈推荐进行基因检测,其阳性率为60-70%。对于非选择性轴突性神经病变,基因检测的阳性率低于10%,但可考虑在排除获得性原因后进行检测。TTR基因引起的淀粉样神经病是一个例外,因有效治疗而备受关注。急性间歇性卟啉病(AIP)通过尿液检测和HMBS/PBGD基因遗传检测进行诊断。
检测方法:
周围神经病可能是多系统遗传疾病的症状之一,其中四种基因(PMP22、MFN2、MPZ、GJB1)占据了90%的诊断率,多基因panel是首选,如不明确,可进行WES/WGS。
临床进展:
已批准的治疗包括Tafamidis和Difunisal用于淀粉样周围神经病,以及Givosiran用于AIP。各种遗传性神经病的潜在治疗方法正在进行临床研究,包括针对SORD基因变异的试验。
九、帕金森病(Parkinson’s Disease)
基因检测的指征和阳性率:
对于任何怀疑帕金森病(PD)的患者,尤其是早发型PD,建议进行基因检测,尤其是有PD、其他运动障碍、痴呆或戈谢病家族史的一级亲属。PD的遗传检测整体上的诊断阳性率为5-26%,包括早发型PD的9-16%和家族性PD的10%。PD基因检测复杂,涉及风险等位基因(如占PD 10%的GBA变异)和低外显率变异(如占PD 2-3%的LRRK2变异)。对于年轻成年人出现帕金森症状,应怀疑威尔逊病,可通过ATP7B基因检测进行诊断。对非典型帕金森性障碍不推荐进行常规基因检测,但DLB可能需要考虑(详见痴呆症)。
检测方法:
建议首先进行基因panel检测,如果结果不明确,则进行WES或WGS。WES/WGS在检测临床相关变异时,可能因附近存在高同源的假基因GBAP1(对GBA基因的干扰)以及SNCA基因易发生结构重排而表现出限制。为克服这一问题,一些帕金森病(PD)的基因芯片采用了专门设计用于检测GBA和SNCA等基因的方法。这种策略有助于提高对这些特定基因变异的敏感性,确保更准确的遗传信息获取。
临床进展:
尽管尚无批准用于遗传PD的治疗方法,基因检测可能会告知个体是否符合参与研究的资格,比如携带GBA或LRRK2变异的患者可参与相应的临床试验。
十、肌张力障碍(Dystonias)
基因检测的指征和阳性率:
Zech等人提出一种外显子测序(WES)算法,建议对符合以下标准之一的患者进行基因检测:20岁前开始、全身或节段性肌张力障碍的患者和伴有其他神经症状。该方法在符合三个标准的患者中具有超过25%的检出率,最高可达50%,而在不符合任意一项标准的患者中,检出率约为1%。
检测方法:
建议首先进行基因panel,如果结果不确定,再进行WES或WGS测序。
临床进展:
尽管尚无特定基因的批准疗法或临床试验,但基因诊断可影响治疗选择,例如对左旋多巴和深脑刺激的响应性。据估计,34%的基因诊断对治疗产生实际影响,包括药物选择、膳食治疗和并发症监测。
参考文献
[1] Dratch L, Azage M, Baldwin A, Johnson K, Paul RA, Bardakjian TM, Michon SC, Amado DA, Baer M, Deik AF, Elman LB, Gonzalez-Alegre P, Guo MH, Hamedani AG, Irwin DJ, Lasker A, Orthmann-Murphy J, Quinn C, Tropea TF, Scherer SS, Ellis CA. Genetic testing in adults with neurologic disorders: indications, approach, and clinical impacts. J Neurol. 2023 Oct 27. doi: 10.1007/s00415-023-12058-6. Epub ahead of print. PMID: 37891417.

 返回搜狐,查看更多
返回搜狐,查看更多
责任编辑:





