Nature(IF:69.5): 环境因素如何塑造肠道菌群
编译:微科盟小木,编辑:微科盟小编、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》公众号。
导读
肠道微生物组与多种疾病相关,但尚未确定健康或不健康微生物群的普遍特征,因此需要了解遗传、暴露、生活方式和饮食如何塑造健康和疾病中的微生物群。本研究分析了来自2756个家庭的三代人群队列中的8208名荷兰人肠道微生物群的细菌组成、功能、抗生素耐药性和毒力因子。我们将这些与241个宿主和环境因素相关联,包括身体和心理健康、药物使用、饮食、社会经济因素以及童年和当前暴露。结果发现微生物群主要由环境和同居塑造。只有约6.6%的分类群是可遗传的,而约48.6%的分类群的变异可以通过同居来解释。通过确定微生物组与健康之间的2856种关联,我们发现看似无关的疾病具有共同的微生物组特征,该特征与合并症无关。此外,我们确定了微生物组特征与饮食、社会经济、早期和当前暴露之间的7519种关联,其中许多早期和当前因素与微生物组功能和组成显著相关。总体而言,本研究全面概述了肠道微生物组以及遗传力和暴露的潜在影响,这将促进微生物群靶向治疗的未来发展。
论文ID
原名:Environmental factors shaping the gut microbiome in a Dutch population
译名:环境因素塑造了荷兰人的肠道微生物群
期刊:Nature
IF:69.504
发表时间:2022.4
通讯作者:傅静远、R. K. Weersma & A. Zhernakova
通讯作者单位:荷兰格罗宁根大学医学中心
DOI:10.1038/s41586-022-04567-7
实验设计
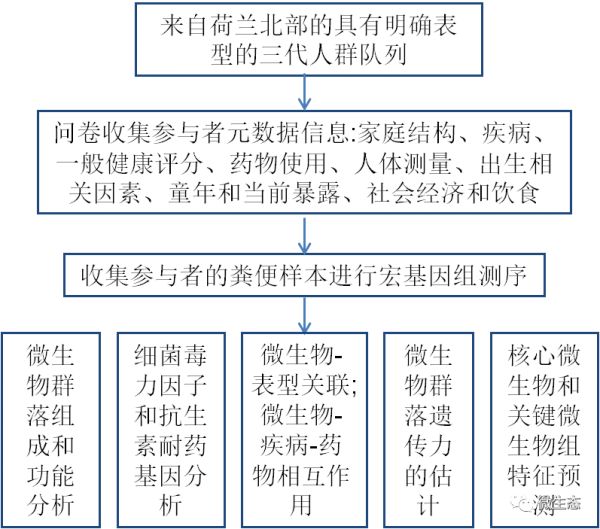
结果
1 荷兰微生物组项目
在Lifelines中启动了荷兰微生物组项目(DMP),这是一个来自荷兰北部的具有明确表型的三代人群队列和生物库。在DMP中,我们描述了8208名个体的肠道微生物群组成和功能(年龄范围8-84岁,57.4%为女性,99.5%为荷兰欧洲血统;其中4745名个体聚集在2756个家庭;图1a,补充表2d)。数据集(由Metaphlan2处理和HUMAnN2)包含1253个分类群(4界,21门,35纲,62目,128科,270属和733种)和564条代谢通路,其中包括257个古菌和细菌分类群以及277条相对丰度高于0.01且存在于超过5%的个体中的通路。
本研究的样本量使我们能够覆盖通过bootstrap分析估计的微生物功能特征总数的90%以上:574.4个(标准误差=4.2)中有564个预期的MetaCyc通路,190个(标准误差= 0.05)中有190个(标准误差=0.05)毒力因子,303个(标准误差=0.03)中有303个(标准误差=0.03)抗生素耐药基因,136.5个(标准误差=2.7)中有128个(标准误差=2.7)科水平或更高水平的微生物分类群(图1b)。通过对队列进行二次抽样,我们估计,当至少对队列的40%(约3300个样本)进行采样时,这些微生物特征的存在率会变得相对稳定(在整个队列观察到的数量的90%以内)。然而,随着样本量的增加,发现的微生物物种数量持续增加,在2.5万份样本中,种群的总物种数量估计为600种(标准误差=27),这表明还有其他稀有微生物物种未被发现(图1b,扩展数据图1)。肠道微生物群组成在种群中高度可变,例如拟杆菌门(Bacteroidetes)的相对丰度在5%-95%以上不等(扩展数据图2c)。与大多数菌门相比,最丰富的微生物通路的可变性明显降低(单侧F检验错误发现率(FDR)<0.05,扩展数据图2d,补充表2g)。
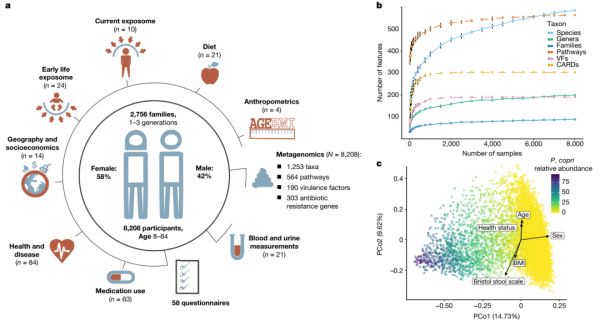
图1 荷兰微生物组项目概述。a,队列的图形摘要和可用元数据的概述(n =收集的变量数,N=样本量)。b,与样本量相关的微生物特征数量。点表示平均值。误差条显示100次重复采样的SD。c, PCoA可视化队列的β-多样性。颜色表示P. copri的相对丰度。箭头表示自我报告的健康、人体测量学和粪便样本元数据的影响。CARDs,来自综合抗生素耐药性数据库的抗生素耐药基因家族;PCo,主坐标;VFs,细菌毒力因子。
2 核心物种
为了找到对肠道生态系统的组织和维持可能至关重要的微生物物种和通路,我们调查了存在于超过95%的个体中的微生物分类群(指定这些核心微生物)和在微生物共丰度网络中形成中心节点的分类群(指定的关键特征)。我们确定了9个核心物种(Subdoligranulum sp.、Alistipes onderdonkii、Alistipes putredinis、Alistipes shahii、单形拟杆菌Bacteroides uniformis、普通拟杆菌Bacteroides vulgatus、直肠真杆菌Eubacterium rectale、Faecalibacterium prausnitzii和Oscillibacter sp.),它们与在英国、美国、欧洲和非西方地区发现的物种高度一致(补充表1a)。我们还确定了28个物种和53条通路作为潜在的关键特征,分别由超过109个和337个显著的共丰度定义(FDR<0.05)。由这些特征定义的网络显示出20.2%的物种重叠和25.3%通路重叠。
9种已确定的核心微生物中的5种(A. putredinis、A. shahii、F. prausnitzii、Oscillibacter sp.和Subdoligranulum sp.)也是关键物种,这意味着它们在荷兰人的肠道微生物生态系统中具有核心作用。例如,F. prausnitzii是一种主要的产丁酸菌,在许多慢性疾病中被消耗,它与大多数拟杆菌(Bacteroidetes)和双歧杆菌属(Bifidobacterium)物种显示出显著的共丰度(补充表1e)。但我们也发现了低流行率(10%)的潜在关键物种,包括Ruminococcus gnavus和梭菌属(Clostridium)的多个物种,它们与多种疾病正相关(补充表3b),这与之前的研究一致。
3 Prevotella copri 定义集群
使用主坐标分析(PCoA)对本研究队列中的微生物组数据进行聚类分析,发现第一个主坐标是由Prevotella copri驱动的(Spearman r = 0.68, P= 3.6×10-180;图1c,补充表9)。该细菌在我们的队列中呈双峰分布,并根据其存在或不存在定义了两个集群(扩展数据图3a, b)。根据之前的研究,我们观察到高丰度的P. copri与较低的肠易激综合征(IBS)风险相关(优势比=0.72,95%置信区间0.61-0.86)。我们还发现P. copri与一般健康水平呈正相关(优势比=1.24,95%置信区间1.11-1.40,FDR<0.05;扩展数据图3c)。尽管之前的研究报道了以拟杆菌属(Bacteroides)、普雷沃菌属(Prevotella)和瘤胃球菌科(Ruminococcaceae)为主的不同肠型,但我们只观察到两个这样的集群,这可能是因为本研究的队列在种族上是统一的,并且来自有限的地理区域。
功能潜力的PCoA显示与许多通路高度相关,而不是单个通路,解释变异的最主要特征是queuosine生物合成、肽聚糖生物合成和L-异亮氨酸生物合成通路(补充表9)。与通路相似,毒力因子的PCoA显示与编码各种细菌功能的多个基因家族相关,包括参与细菌侵袭肠道细胞的鞭毛蛋白(基因家族VF0114,Spearman r = 0.60,P≈0.0)、细菌铁载体(VF0136、VF0228和VF0256;Spearman r = 0.55、0.58、0.51;P <1.0×10-100)、分泌系统(VF0333;Spearman r=0.76,P≈0.0)和细菌粘附因子(VF0221和VF0404;Spearman r=0.581和0.561;P<1.0×10-100)。相比之下,抗生素耐药基因的PCoA显示主要由3个对四环素抗生素产生耐药性的基因家族组成:核糖体保护蛋白ARO_30001914和ARO_3000191编码基因家族(Spearman r分别为0.81和0.68;P≈0.0)和外排泵编码基因家族ARO_3000567 (Spearman r=0.50,P≈0.0)。
4 同居胜过遗传力
接下来,我们探讨了家庭结构、同居和其他暴露因素在肠道微生物群形成中的相对贡献。使用研究队列的多代家族结构来估计微生物类群的遗传力,并在经FDR校正的P<0.1时确定了17个可遗传类群(占测试类群的6.6%)(图2a,补充表5)。变形菌(Proteobacteria)的遗传力最高(h2=0.308,其中h=狭义遗传力(由加性遗传效应引起的微生物分类群丰度的变化比例)),其次是嗜黏蛋白阿克曼氏菌 (Akkermansia muciniphila,h2=0.302)及更高水平的类群拟杆菌科(Bacteroidaceae,h2=0.299),拟杆菌科物种Parabacteroides goldsteinii(h2=0.266)和Bacteroides coprocola(h2=0.228)、长双歧杆菌(Bifidobacterium longum,h2=0.247)、Phascolarctobacterium属(h2=0.245)和梭菌目(Clostridiales)的属水平集群(h2=0.237)。在微生物通路中,只有7个在经FDR校正的P值<0.1时是可遗传的,包括脂质IVA生物合成通路(NAGLIPASYN-PWY)、5-磷酸吡哆醛生物合成的两条通路(PWY0-845和PYRIDOXSYN-PWY)、异亮氨酸生物合成II通路(PWY-5101)和前醌生物合成通路(PWY-6703)。通路和分类群的遗传力表现出一定程度的一致性:NAGLIPASYN-PWY与变形菌高度相关(R=0.64),其他可遗传通路大多与拟杆菌科(Bacteroidaceae)和部分拟杆菌科物种相关。
之前已观察到其中一些分类群的遗传力。据报道,在对英国双胞胎的研究中,阿克曼氏菌(Akkermansia)和双歧杆菌(Bifidobacterium)以及梭菌目中的一些属是可遗传的,并且在加拿大人群基于家族的分析中报告了拟杆菌科成员的遗传力,包括拟杆菌属(Bacteroides)和副拟杆菌属(Parabacteroides)。我们的结果并没有复制这些研究中的一些可遗传分类群,这可能是由于所使用的技术(使用16S和宏基因组测序以及不同的DNA分离方法)或参考数据库的差异,即一些类群不在参考数据库中(如克里斯滕森菌科Christensenellaceae在Metaphlan2数据库中),没有超过5%的存在阈值(Turicibacter),或者由于存在率低而显示出非常低的遗传力检测能力(利用Metaphlan3数据库分析Christensenellaceae、Euryarchaeota)。
同居的影响比遗传的影响大得多,257个分类群中有125个(48.6%)受到同居的显著影响,同居史解释了22个类群(8.6%)的显著差异(补充表5a)。无论参与者的亲缘关系如何,同居参与者的微生物组也比分开生活的参与者更相似,父母子女组、兄弟姐妹组和不相关伙伴的微生物组比非同居参与者的微生物组更相似(图2c-e,基于Wilcoxon检验FDR<1.0×10-5)。我们进一步观察到微生物通路、毒力因子和抗生素耐药基因组成的相似模式(扩展数据图4a-c)。
这些结果表明,整个微生物组的组成受到同居的显著影响,而遗传的作用较小,虽然少数微生物类群(如A. muciniphila、B. longum和Bacteriodaceae)具有显著的可遗传性并在不同群体间表现出一定程度的复制。但考虑到本研究队列(方法)的设置以及在几个分类群和通路上观察到的比较高的同居效应,我们对遗传力的估计可能会略微夸大。
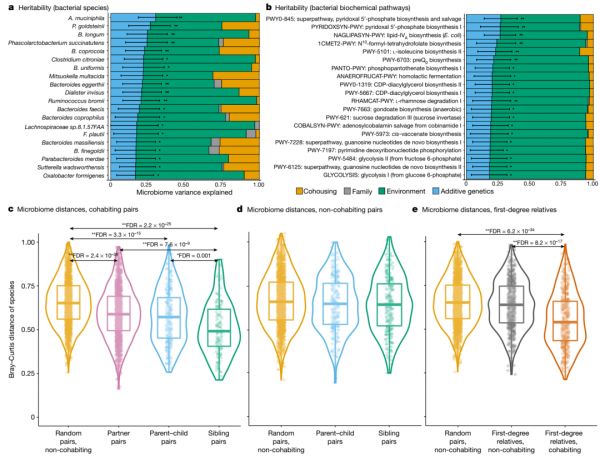
图2 遗传力及同居对肠道微生物群的影响。a, 20个可遗传物种。b,前20条可遗传通路。**FDR < 0.1时,可显著遗传的分类群;*具有名义上显著遗传力的分类群(P < 0.05)。误差线显示遗传力的95%置信区间。a,b的结果是从1432个家庭的3571个不同个体中计算出来的。c-e,成对微生物组距离比较。中心线是中位数,框限表示上、下四分位数,须表示1.5×四分位数范围,点表示异常值,轮廓显示数据的分布。通过将随机非同居组(n=2000)与同居伴侣(n =1710)、父母子女组(n=285)和兄弟姐妹组(n=144) (c),随机配对(n=2000)与非同居父母子女组(n=301)和兄弟姐妹组(n=299) (d)以及随机配对(n=2000)与非同居一级亲属组(n=600)和同居一级亲属对(n=429) (e)的微生物物种进行比较来计算Bray-Curtis相异度,在c-e中,**FDR<1.0×10-5,*FDR<0.05(基于Wilcoxon检验,双侧)。
5 关联概述
然后,我们探索了微生物特征与241项测量指标的关系,包括技术因素、人体测量、早期和当前暴露、饮食、自我报告的疾病和药物使用、医学测量和社会经济(扩展数据图5)。这些表型解释了12.9%的微生物组分类组成和16.3%的微生物组功能,其中最大的贡献来自技术因素、粪便特征、疾病、药物使用和人体测量(图3b,补充表4a-c)。
校正技术因素后,我们观察到4530个表型与分类群相关,5224个表型与通路相关,1848个表型与抗生素耐药基因相关,385个表型与毒力因子相关(补充表3g,扩展数据图5)。单独而言,与关键类群和核心类群(Flavonifractor salivarius、F. prausnitzii、Alistipes senegalensis、Clostridium和Subdogranulum物种)的关联数量最大(补充表3b, h)。A. senegalensi以前与克罗恩病和乙型肝炎病毒相关的急性慢性肝功能衰竭有关。在本研究队列中,A. senegalensi与43个表型相关(FDR < 0.05),突出了其在多种疾病中的潜在作用。同样,与2型糖尿病(T2D)、高血压和强直性脊柱炎相关的Clostridium asparagiforme与另外23种不相关的疾病有关(FDR < 0.05)。下面讨论了进一步的关联,补充表3a-h提供了完整概述。
在β-多样性的个体间变异分析中,年龄、性别和BMI是最主要的表型,分别解释了0.6%、0.53%和0.32%的个体变异。Bristol粪便量表解释了最大比例的β多样性(R2 =0.77%,FDR=0.012),采样季节也解释了很大比例的变异(R2 =0.36%,FDR=0.012,补充表4a),它们共同强调了在微生物组研究中评估粪便稠度和收集时间框架效应的重要性。在这些结果的基础上,我们将年龄、性别、BMI、Bristol粪便量表和采样季节的校正与技术因素的校正一起纳入我们的关联模型(补充表3a-e)。
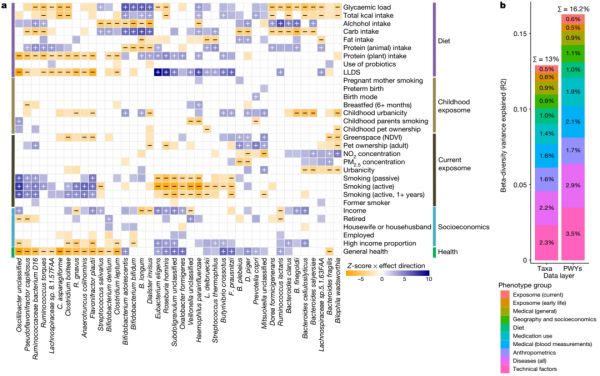
图3 微生物-表型关联。a,与健康微生物组特征相比,饮食、童年和当前暴露以及社会经济的微生物-表型关联。关联数量最多的前40个微生物物种通过关联Z分数使用层次聚类法进行聚类,并按效应方向着色(蓝色,正;橙色,负),在整个研究范围内FDR <0.05,正相关为加号,负相关为减号。没有标记的颜色关联表示名义上显著的关联(P < 0.05)。b,在多变量PERMANOVA分析中,表型组解释了微生物组组成和功能的差异。NDVI,标准化植被指数;PWY,通路。
6 健康和不健康微生物的定义
为了定义健康和疾病的微生物组特征,我们将微生物组特征与自我报告的健康和至少20例的81种疾病相关联(补充表2d)。确定了1206个与细菌分类群的显著关联,1182个与微生物通路的关联,390个与抗生素耐药基因的关联,76个与细菌毒力因子的关联(FDR<0.05,补充表3b-f)。不同疾病有不同数量的关联,在心血管和代谢疾病(如非酒精性脂肪肝和T2D)以及胃肠道疾病(包括炎症性肠病和肠易激综合征)中观察到最强的特征(补充表3f)。在大多数疾病中观察到一致的微生物组-疾病模式(图4a),这使我们能够查明不相关疾病之间共享的微生物组特征以及定义健康(即无疾病)微生物组的特征。
疾病的共同微生物组特征主要包括厌氧菌属(Anaerotruncus)、瘤胃球菌属(Ruminococcus)、拟杆菌属(Bacteroides)、Holdemania、黄杆菌属(Flavonifractor)、Eggerthella和梭菌属(Clostridium)物种增加以及粪杆菌属(Faecalibacterium)、双歧杆菌属(Bifidobacterium)、丁酸弧菌属(Butyrivibrio)、Subdoligranulum、草酸杆菌属(Oxalobacter)、真杆菌属(Eubacterium)和罗斯氏菌属(Roseburia)减少。不相关疾病共有的肠道微生物组通路主要包括L-鸟氨酸、泛醇和甲基萘醌、肠杆菌共同抗原、Kdo-2-lipid-A和钼辅因子的生物合成增加,以及氨基酸、脱氧核糖核苷和核苷酸的生物合成、厌氧能量代谢和发酵产生短链脂肪酸(主要是丁酸)减少。某些疾病的毒力因子增加,包括T2D和胃肠道疾病,其中细菌粘附和铁摄取因子(VF036、VF0228、VF0236、VF0404和VF0394)的影响最大。通过构建L1/L2正则化回归模型对36种最常见疾病的预测进一步验证了这些特征,并发现与关联分析具有高度一致性,模型选择的31个物种中有22个与超过5种疾病相关(补充讨论)。尽管本研究队列中的疾病合并症较低,但预测特征显示出高相关性(图4b,补充表6e)。
最后,我们计算了最近开发的肠道微生物组健康指数(GMHI),发现健康个体和疾病个体之间存在显著差异(P=6.16×10-22;扩展数据图6)。我们的健康微生物组特征在属或种水平上复制了50个GMHI特征中的43个(补充表10)。此外,我们确定了GMHI研究中未发现的55种微生物组-健康关联,包括来自丁酸弧菌属(Butyrivibrio)、阿克曼氏菌(Akkermansia)和普雷沃菌属(Prevotella)的物种(补充表3b, 10),这些物种此前与胃肠道疾病有关,但与其他疾病无关。
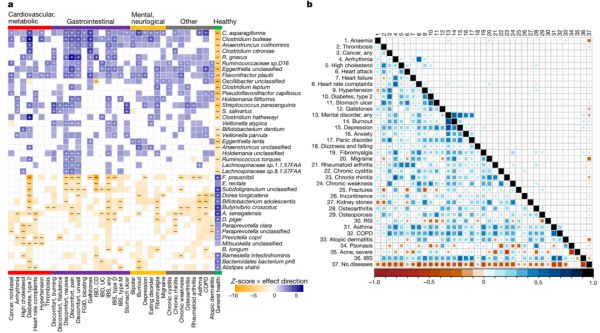
图4 健康和疾病的微生物组特征。a ,与疾病类别和健康状况相关的微生物物种的热图。疾病按疾病类型分类和标记。关联数量最多的前40个微生物物种通过关联 Z 分数(由颜色强度表示)使用层次聚类法进行聚类。关联按效应方向着色(蓝色,正;橙色,负),在整个研究范围内FDR < 0.05时关联显著,正相关和负相关分别用加号和减号标记。没有标记的彩色关联表示名义上显著的关联( P < 0.05,未通过多重检验校正)。 b ,队列中预测疾病的特征(左下三角)与这些疾病的合并症(右上三角)之间的Pearson相关性比较。CD,克罗恩病;COPD,慢性阻塞性肺病;IBD,炎症性肠病;IBS,肠易激综合征;RSI,重复性劳损;UC,溃疡性结肠炎。
7 微生物组反映疾病和治疗
我们观察到,常见非传染性疾病的微生物组效应与用于治疗这些疾病的药物具有高度一致性(扩展数据图7,补充表3a-g)。在这些药物中,我们发现质子泵抑制剂(PPIs)、抗生素、双胍类抗糖尿病药、渗透性泻药和肠道抗炎药的作用最强(分别与微生物类群有84、56、47和32个关联,FDR<0.05)。为了阐明疾病和药物的影响,我们对三种常见疾病及其相应的常见药物进行了多变量线性回归分析:T2D中使用的抗糖尿病药物、抑郁症中使用的选择性5-羟色胺再摄取抑制剂(SSRIs)以及功能性胃肠道疾病和IBS中使用的PPIs。我们观察到,疾病和相应药物的作用与微生物组具有高度一致性,即使在相互制约的情况下也是如此(补充表7),这表明不健康的肠道微生物组特征反映了疾病和相关药物。
8 儿童期与成年期微生物组有关
由于生命最初的2-3年对微生物群的发育至关重要,我们研究了生命早期(小于4岁)因素对成人微生物群的影响。我们确定了106个与分类群的关联,30个与通路的关联,22个与抗生素耐药基因的关联,2个与毒性因子的关联(FDR<0.05),仅观察到对分娩方式、母乳喂养和早产的影响很小(图3b,扩展数据图8)。尽管儿童和成人城市化之间的相关性非常低(Spearman r<0.015),但童年生活环境与成人微生物组显著相关(FDR<0.05时,分别有54、8和7个分别与分类群、通路和CARD相关)。
农村童年环境与各种细菌的增加有关,包括P. copri、F. prausnitzii、Rothia mucilaginosa以及来自双歧杆菌属(Bifidobacterium)和Mitsuokella的物种,它们的丰度也与总体健康状况的增加有关。相比之下,在城市童年环境的参与者中,拟杆菌属、Alistipes和Biophila的多个物种的丰度降低。
父母吸烟与其子女的微生物组相关(FDR<0.05时,分别有15、9和4个与分类群、通路和CARD相关)。在本研究中,我们观察到父母吸烟与韦荣氏菌属(Veillonella)和Oscillibacter属物种以及P. copri丰度降低之间的关联(R2= 0.0012,FDR=0.033),这与当前吸烟者的观察结果一致。最后,童年时期养宠物与成年后的微生物组有关(FDR<0.05时有7个关联),如在童年养过宠物的参与者中观察到Alistipes finegoldii、Lactobacillus delbrueckii、小类杆菌属(Dialister)和嗜胆菌属(Bilophila)物种减少。
9 暴露体与肠道微生物组有关
研究了采样时的环境因素,并确定了健康微生物组特征与宠物饲养、农村生活环境和生活环境绿地表面积的共同关联模式(图3b),包括P. copri、Bacteroides plebeius、Desulfovibrio piger和Mitsuokella属物种增加,脆弱拟杆菌(Bacteroides fragilis)和Bilophila wadsworthia减少。这些特征与增加的NO2和小颗粒物污染物测量值的微生物组关联形成对比,后者与健康呈负相关(扩展数据图9)。吸烟表型,包括当前的主动、被动吸烟以及吸烟史,显示出一致的微生物组关联方向,这也与微生物组-疾病关联的特征相匹配(扩展数据图9)。主动吸烟与41个物种和84种通路有关(FDR<0.05,图3b),其中60%也与吸烟史有关,表明吸烟具有长期影响。值得注意的是,其中15种也与被动吸烟有关,这强调了在疾病风险模型中考虑被动吸烟的必要性。
我们观察到社会经济因素(如月收入和社区收入)与肠道微生物组之间的220个关联(FDR<0.05),其中72个为细菌丰度和月收入之间的关联;较高的收入与健康的微生物组特征相关。收入与社区绿地面积、农村生活环境和生命线饮食评分(LLDS)的相关性虽低但显著(Spearman相关系数分别为0.22、0.17和0.07;相关检验FDRs< 1.0×10-6),这意味着微生物组-收入关联可能反映了多种因素的组合,包括更健康的饮食和生活方式以及较少城市化的生活环境。这些结果支持肠道微生物组可能是整个社会经济阶层中众所周知的健康差异的中介的假设。
本研究确定了20种饮食因素和82个物种之间的378种关联(补充表3b)。在食物频率问卷(FFQ)收集和粪便采样之间的5年期间,饮食也相对稳定,代表主要饮食项目和饮食趋势的30个FFQ项目在测量中保留超过95%,其余项目保留超过50%(补充表8,补充讨论)。
LLDS是基于国际营养文献的饮食评分,显示出最多的关联(79个与分类群相关,44个与通路相关,20个与抗生素耐药基因相关,8个与毒力因子相关,FDR<0.05),其次是总酒精摄入量、血糖负荷、蛋白质饮食评分(反映蛋白质的数量和来源)和总碳水化合物摄入量。LLDS和蛋白质摄入评分显示出与一般健康状况增加的微生物组特征重叠的关联模式,例如梭菌属(Clostridia)物种减少,丁酸弧菌属(Butyrivibrio)和罗氏菌属(Roseburia)以及参与泛醇和甲基萘醌合成的通路增加。相比之下,总膳食碳水化合物摄入量和血糖负荷显示出相反的关联(图3b,扩展数据图10)。
讨论
除了确认已知的微生物组与年龄、性别和BMI的关系外,我们的结果强调了经常被忽略的与粪便样本(粪便稠度)、采样季节和样本处理(DNA浓度或测序批次)相关的混淆因素的重要性。这在研究年龄、性别、BMI和粪便稠度与疾病相关的疾病或探索具有季节性变化的表型(如饮食、身体活动和过敏、流感、普通感冒等疾病)时尤为重要。
我们观察到微生物组主要与同居和环境有关,而非遗传相关性,这证实了之前的研究,即确定有限的整体微生物组遗传力、同居家庭成员及其宠物之间共有的微生物组模式以及停止同居的双胞胎的微生物差异。这些结果表明,遗传力较低的细菌(如Ruminococcus、Streptococcus和Veillonella)可能比遗传力更强的细菌(如Akkermansia、Collinsella和Bacteroides)更容易受到微生物群落改变的影响。
通过比较微生物组、健康和多种疾病之间的关联,我们发现了肠道生态失调的共同特征(图4a),这与之前的研究基本一致。共有的生态失调的存在对微生物组研究和微生物群靶向诊断和治疗具有重要意义。正如我们的预测模型和以前的研究所支持的那样,共有的生态失调意味着肠道微生物群是一般健康的生物标志物,但也使基于微生物群的个别疾病诊断复杂化。由于单一疾病模型可能会被不相关疾病之间共有的特征所混淆,因此在混合疾病队列中测试这些模型的特异性将是临床实施前的重要一步。共同的微生物组特征还表明,微生物组靶向干预措施可以改善人类整体健康。我们的观察结果支持了这一点,即通常认为健康的生活方式因素(例如坚持当前的饮食建议和不吸烟)与一般健康相关的微生物组模式相关。尽管微生物组-药物相互作用已经在体外得到了很好的描述,并且已经在体内对抗生素、PPIs和抗糖尿病药进行了表征,但我们的结果表明,一般微生物群失调是药物和疾病效应共同作用的结果,这意味着许多目前研究不足的药物(如SSRIs)可能会对肠道微生物组产生负面影响。
未报告疾病或药物使用的人群参与者中存在疾病样微生物组特征,表明人群中存在临床前隐性疾病,并表明肠道菌群失调可能先于慢性疾病(如T2D)的临床发作。虽然这一假设需要对长期纵向队列进行实验验证或分析,但我们的观察结果表明,肠道微生物群可用于监测长期健康状况并检测临床前阶段的疾病。
将健康和不健康的微生物组模式与童年和当前暴露、饮食和社会经济联系起来,观察到更健康的饮食、童年和当前对农村环境和宠物的接触、接触绿地以及更高的收入与健康的微生物组模式共享特征。这些观察结果支持微生物群落多样性假说(也称为卫生假说),该假说认为,减少微生物暴露有助于增加自身免疫和过敏性疾病的频率。值得注意的是,尽管经典的卫生假说侧重于病原菌和生命早期的暴露,但我们的结果表明,成人暴露也会导致健康或不健康的微生物组模式,并且环境塑造了整个生命周期中的微生物组,这意味着微生物群靶向疗法可能在个体的整个生命周期都是有效的。此外,我们发现饮食评分、宠物和农村环境与条件性病原菌(如梭菌属)呈负相关,而与共生病(如拟杆菌属、Alistipes和粪杆菌属的产丁酸菌)呈正相关,这意味着暴露于环境中的病原菌和共生菌对建立健康的肠道生态系统具有重要作用。
本研究还观察到吸烟、高碳水化合物饮食以及暴露于NO2和小颗粒物(PM2.5)和与疾病相关的梭菌属和瘤胃球菌属物种呈正相关。尽管空气污染物与肠易激综合征(IBS)等胃肠道疾病有关,并已被证明会影响小鼠的肠道微生物群,但它们对人类肠道微生物群的影响在很大程度上仍未被探索。我们的研究结果与之前对美国队列中空气污染关联的分析一致,表明空气污染物对人类肠道微生物群产生负面影响,并可能通过导致一般的生态失调而增加胃肠道疾病的风险。我们的观察结果进一步证实了这一点,即IBS与PM2.5污染物相关(Spearman r = 0.15),而污染物与其他疾病、社会经济因素和饮食之间的相关性非常低(Spearman r <0.05)(补充表2c)。
本研究发现,儿童时期接触吸烟、宠物和农村环境与成人微生物组有关。尽管这些关联的影响大小低于当前暴露,但影响方向和模式是一致的,这表明环境暴露可以产生长期影响,而且微生物组反映了个体的暴露史。我们的发现进一步支持了这一点,即曾吸烟者仍然显示出与当前吸烟者相似的微生物组关联,尽管影响较小。
测量了241种广泛的表型,但我们只能解释大约15%的微生物组组成和功能的个体间变异,这与之前的大规模研究一致。这意味着肠道微生物组是高度个体化的,包含可能难以从数据中的人工制品中分离出来的稀有类群,而且我们目前对塑造肠道微生物组的因素的理解仍然有限。这种低解释力也可能反映了以数据库为中心的微生物组分类的使用,这种分类在促进研究标准化和低假阳性率的同时,排除了对未分类微生物的识别。未来可能通过基于组装和独立于数据库的方法和纵向研究来量化“缺失变异”,这将在微生物组靶向诊断和治疗的开发中发挥关键作用。
结论
本研究生成并分析了一个大型的、多代的肠道微生物组队列,并将其与广泛的表型数据相关联。我们定义并描述了多种疾病共有的肠道生态失调,并确定了这种生态失调与遗传力、童年和当前暴露、生活方式和社会经济之间的联系。本研究展示了大规模、表型良好的队列在剖析肠道微生物组、健康、遗传和环境之间的联系方面的力量,并为未来微生物组干预的研究提供了丰富的资源。
本文由“健康号”用户上传、授权发布,以上内容(含文字、图片、视频)不代表健康界立场。“健康号”系信息发布平台,仅提供信息存储服务,如有转载、侵权等任何问题,请联系健康界(jkh@hmkx.cn)处理。
相关知识
Nature首次证实:节食会改变肠道菌群组成,增加致病菌
Nature 重磅研究:剖宫产会严重影响婴儿肠菌!微生物多来自医院,是潜在致病菌
Nature子刊:移植瘦子的肠道菌群,能够改善肥胖患者健康
Nature子刊:膳食纤维促进肠道细菌对有益氨基酸的转化
猫狗肠道菌群—“主子们”的健康新领域
人肠道产甲烷菌与肠道健康
健康新知|低碳饮食减重的决定因素竟然是肠道菌群?
个体肠道菌群是精准营养干预代谢健康成功的基础
爱幼妈妈:肠道微生物与婴幼儿健康研究进展
顶刊综述丨NAT REV MICROBIOL (IF:78): 胆汁酸和肠道微生物群: 代谢相互作用和对疾病的影响
网址: Nature(IF:69.5): 环境因素如何塑造肠道菌群 https://www.trfsz.com/newsview90358.html
推荐资讯




- 1发朋友圈对老公彻底失望的心情 12775
- 2BMI体重指数计算公式是什么 11235
- 3补肾吃什么 补肾最佳食物推荐 11199
- 4性生活姿势有哪些 盘点夫妻性 10428
- 5BMI正常值范围一般是多少? 10137
- 6在线基础代谢率(BMR)计算 9652
- 7一边做饭一边躁狂怎么办 9138
- 8从出汗看健康 出汗透露你的健 9063
- 9早上怎么喝水最健康? 8613
- 10五大原因危害女性健康 如何保 7828

